Modelling
The modelling pipeline trains drug-disease treatment prediction models using indication/contraindication data and knowledge graph embeddings.
Overview
The modelling pipeline implements a robust cross-validation strategy with ensemble learning to predict whether a drug treats a disease. The pipeline classifies three categories of drug-disease relationships:
- Treat: Positive relationships where a drug treats disease, represented by known indications.
- Not Treat: Negative relationship, represented by known contraindications.
- Unknown: Negative relationship, represented by random drug-disease pairs where the drug is not related to the disease.
For every drug disease pair, we compute scores for all 3 classes. The scores add up to 1.
Pipeline overview
The modelling pipeline follows a systematic approach to train robust prediction models. This is described as follows where we fix several concrete hyperparameters for simplicity.
Note
All of these stages run for every model listed in matrix.settings.DYNAMIC_PIPELINES_MAPPING()["modelling"]. Shared steps (splits, negative sampling) execute once, after which model-specific transformers, estimators, and ensembles are produced per fold. See the Multi-Model Configuration Guide for configuration details, parameter sources, and downstream aggregation behaviour.
-
Cross-Validation Setup:
-
Folds 0, 1, 2: Training folds with different test/train splits (90%/10%) for known positives and negatives.
-
Fold 3: Full training data fold (no test split) for final model production
-
Shard Generation: For each fold, create 3 shards with different randomly sampled negative examples
-
Model Training: Train individual classifiers on each fold-shard combination using the transformed feature data
-
Ensemble Creation: Combine predictions from all shards within each fold using aggregation functions to produce one ensemble model per fold
-
Production Model: Use the Fold 3 ensemble model (trained on full dataset) for final matrix generation and inference
You can find further details for each step below.
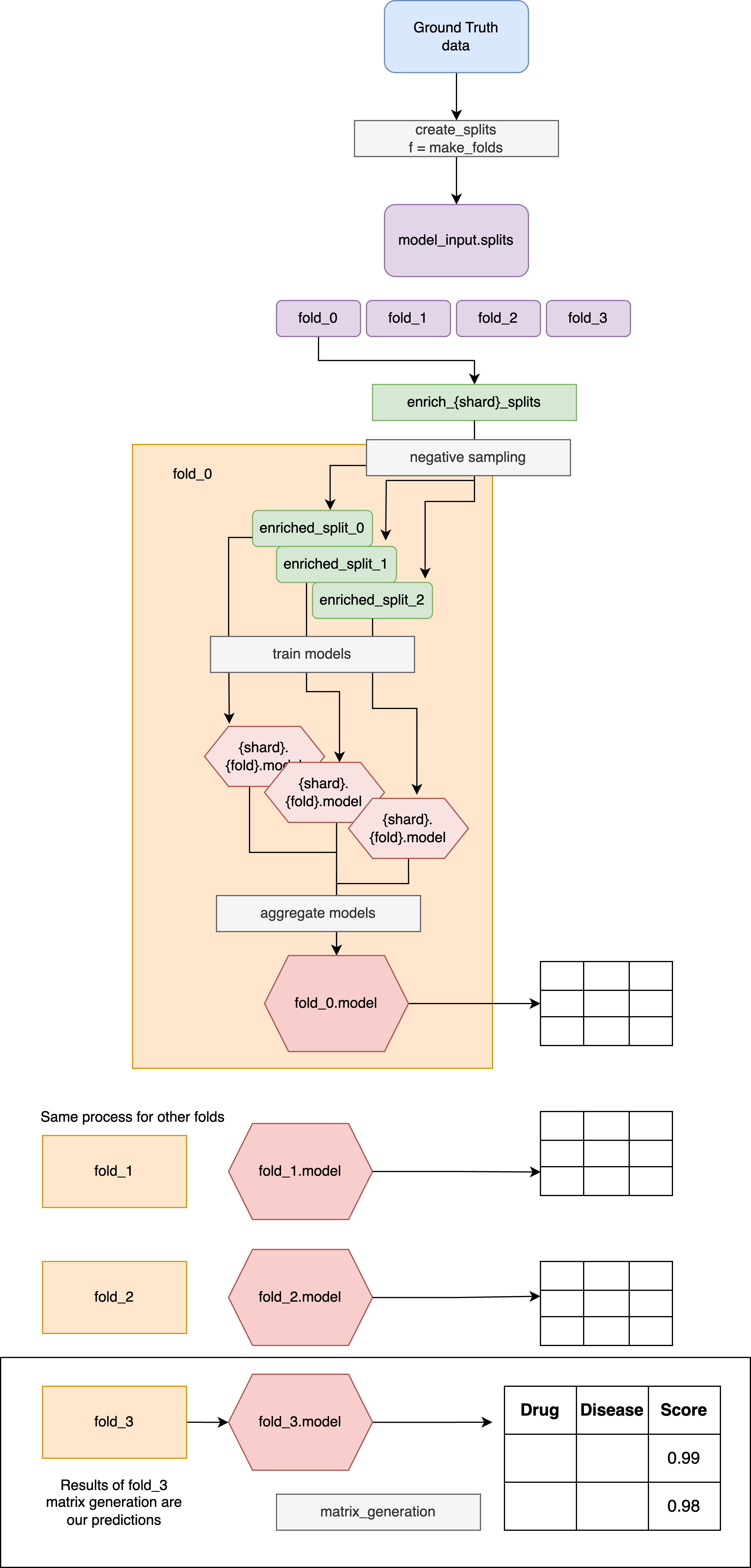
Pipeline steps
1. Cross-Validation Setup
The pipeline uses a cross-validation approach with multiple folds. We can adjust the number of folds in the settings. The same fold boundaries are shared across all configured models, and fold-specific outputs are namespaced by model (e.g. modelling.fold_{fold}.{model_name}.models.model).
- Folds 0 to n-1: Training folds used for model evaluation
- Each fold contains different train/test splits from the ground truth data
- Used to assess model performance and prevent overfitting
- Fold n: Full training data fold used for final model training
- Contains all available training data (no test split)
Info
Note that it is always the last fold that is trained on the full data, For example, in case of a 3-fold model, fold_0, fold_1 and fold_2 represent the 3 cross-validation training folds. fold_3 is the model used to generate results that are shared with the Medical Team.
If we used 5 cross-validation folds, fold_5 would be the fold we use to generate the full results.
Ground Truth Data
The pipeline pulls in ground truth data from the release containing known positive and negative examples from different sources (e.g. KGML-xDTD, EC Indications). As of August 2025, the data releases contain a unified ground truth product which concatenates different training sets that EveryCure considers valuable. Using this unified ground truth set, you can now select which Ground Truth you want to use for model training by specifying sources in defaults.yaml configuration for modelling pipeline.
For instance, the following parameters specify that all datasets (ec, drugbank, kgml_xdtd) should be chosen for training e.g.
modelling.training_data_sources:
- ec # EveryCure Indications
- kgml_xdtd # KGML-xDTD Indications
Configuration that can be found on main branch for matrix is our most up-to-date which leads to the most performing drug repurposing predictions.
Please see the data documentation for more information on the datasets.
Split Generation
The make_folds function creates cross-validation splits using a defined splitter (e.g. Disease split, KFold).
For example, for a 5-fold cross-validation strategy ensures that each ground truth pair appears in the test split of exactly one fold (repeated per model configured in the dynamic mapping):
- Generates train/test splits for folds 0-4 using the specified splitter strategy
- Train ~80%
- Test ~20%
- Creates a full training dataset for fold 5 (no test split)
Note
The number of folds can be adjusted in the configuration settings.
Hyperparameter Optimization Process
For any fold, the pipeline follows this hyperparameter optimization strategy:
- Split Training Data: Further split the training data into strict train and validation sets
- Hyperparameter Search: Find optimal hyperparameters by training on strict train and evaluating on validation
- Final Training: Train the model with optimal hyperparameters on the full training set
2. Shard Generation - Negative Sampling Implementation
The pipeline implements a negative sampling approach to create training data for the "unknown" class.
We create \(n\) shards as defined in the settings, each with a different randomly sampled set of negative examples. Multiple shards help reduce the impact of random sampling bias on model performance. Shards are generated independently for every model so that the tuned estimator follows the negative-sampling strategy specified in that model’s configuration file.
Negative sampling is implemented by:
- Taking a known positive drug-disease pair
- Extracting either the drug or disease node from the pair
- Replacing it with a randomly sampled node from the respective drug or disease pool
- The drug and disease pools are defined in the configuration
This approach ensures that the negative samples maintain realistic drug and disease entities while creating pairs that are unlikely to have treatment relationships.
Note
Negative samples serve as the sole source of data for the "unknown class". They are essential to ensure that the training dataset represents the vast majority of pairs, which are unrelated to each other.
3. Model Training
For each shard in each fold, the pipeline trains a classifier on the transformed feature data. The pipeline supports any scikit-learn BaseEstimator object including Random Forest, XGBoost, and Fully Connected Neural Networks (FCNN). Trainers, transformers, and feature selections are supplied per model via the configuration files referenced in the multi-model guide.
4. Ensemble Creation
After training individual shard models, the pipeline creates ensemble models:
- Combines shard-level estimators within each fold using the aggregation function configured for that model.
- Produces a single
ModelWrapperper fold and per model that encapsulates the aggregated estimators. - Applies the ensemble wrapper to generate predictions on the validation/test split so performance can be monitored.
Matrix generation packages each ensemble with its preprocessing (ModelWithTransformer) and aggregates across the configured model types before scoring; refer to the Multi-Model Configuration Guide for how those wrappers are built, combined, or overridden.
Multi-Model Configuration
The modelling pipeline supports training multiple model types in parallel (e.g., XGBoost variants, LightGBM). Matrix generation then packages each trained model with its preprocessing and aggregates predictions across models.
For setup, per-model parameters, and aggregation details, see the Multi-Model Configuration Guide.